Guidelines to
HYDROPHOBIC CLUSTER ANALYSIS
(HCA)
The HCA method is
based on the use of a bidimensional plot, called the HCA plot, the
principles of which are illustrated below (Figure
1).
The bidimensional plot originates
from the drawing of the 1D sequence on an an alpha helix (3.6
residue/turn, connectivity distance of 4 (residues separating two
different clusters) which has been shown to offer the best
correspondence between clusters and regular secondary structures.
Examination of the HCA plot of a protein sequence allow to easily
identify globular regions from non globular ones and, in globular
regions, to identify secondary structures. This 2D signature, which is
much more conserved than 1D sequence and which can be enriched from the
comparison of families of highly divergent sequences, allows to
succesfully detect at low levels of sequence identity relevant
similarities.
For more details about the methodology and applications, see our publications.
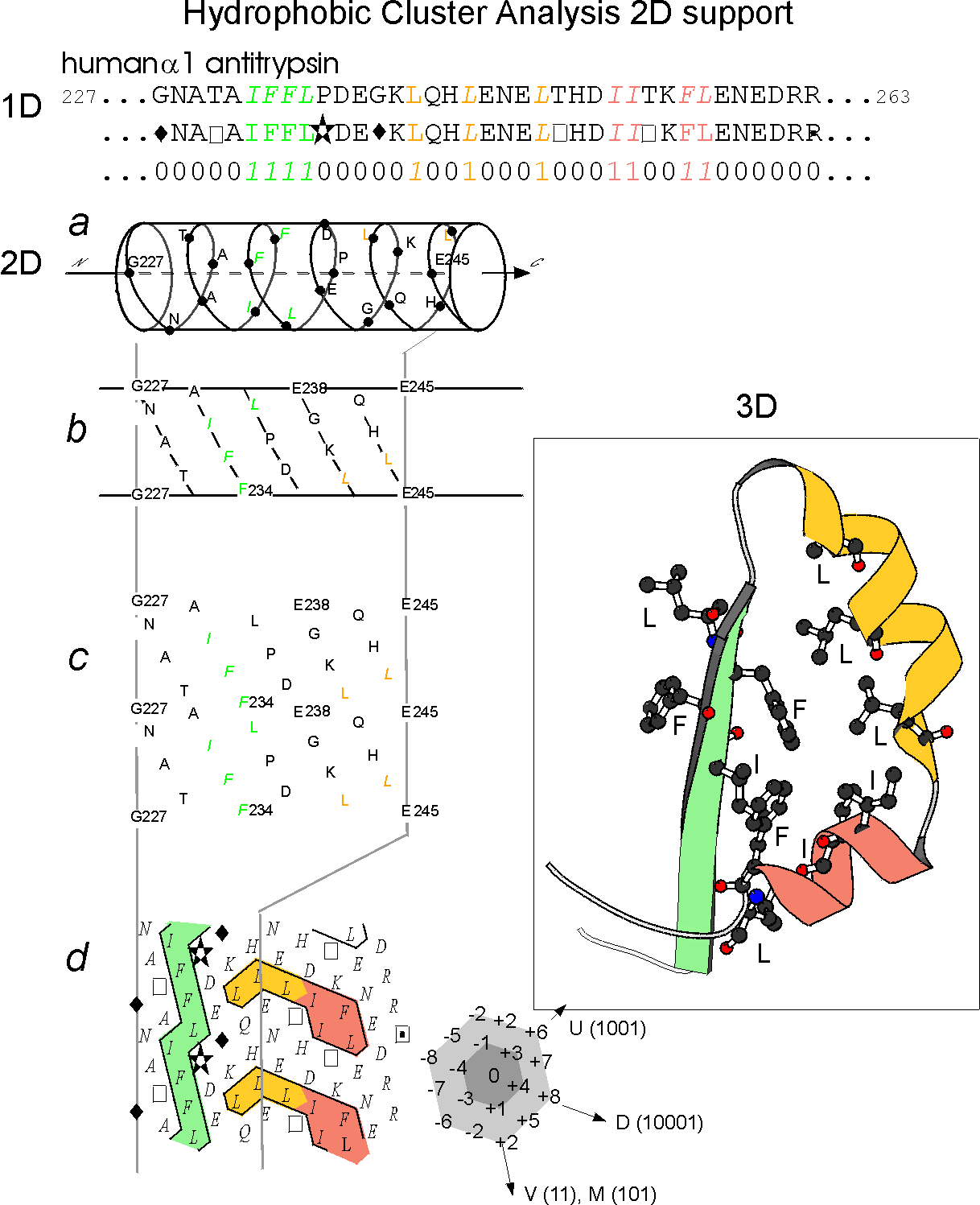
Figure 1 (adapted from the figure 1 of Ref.1)
Illustration of the
principles of the HCA diagram
The protein
linear sequence (1D) (here the human alpha1 antitrypsin) is shown on
the top of the figure with hydrophobic amino acids coloured. This
sequence is written on an alpha helix displayed along a cylinder.
The cylinder is then cut parallel to its axis and unrolled in a
bidimensional diagram (2D). This diagram is compacted and duplicated in
order to restore the full environment of each amino acids. Hydrophobic
amino acids are not distributed random but form clusters. The positions
of these clusters have been shown to correspond to the positions of
regular secondary structures (alpha helices and beta strands). This is
illustrated by the correponding experimental structure (3D). The form
of the clusters is generally indicative of the type of secondary
structures (vertical clusters are often associated to beta strands
whereas horizontal ones often correspond to alpha helices).
Special symbols are used for some amino acids: star for proline, square
and dotted square for threonine and serine and diamond for glycine.
A detailled list of the percentages
of alpha, beta and coil structures associated to each cluster (as
deduced from experimental structures) is in preparation. Conversely,
sequences stretches between clusters mainly correspond to loops. The 2D
structure of a protein sequence can be therefore easily deduced from
the examination of the HCA plot.